Cells are the fundamental units of life, with a complex organization that allows them to perform diverse and specialized functions essential for the survival and operation of multicellular organisms. Despite the wide range of shapes, sizes, and specific roles cells may have, they share several key characteristics that underlie their functionality and adaptability. These characteristics include their structural organization, surface interactions, motility, internal organization, and mechanisms of communication with other cells and the external environment. Through highly regulated processes, cells maintain balance within their internal environment while engaging in constant exchanges and responses to external stimuli. Here, we examine the general characteristics of cells, detailing their surface structure, movement, membrane organization, gene regulation, protein synthesis, intercellular communication, and size variations. This overview provides insight into the intricate ways cells support both individual cellular function and the larger systems within which they operate.
1. Cell Surface and Size
- Surface Interaction: Cells interact with each other, their environment, and internal structures, often having folded surfaces, especially in cells involved in absorption or transport, such as epithelial cells.
- Size Limitation by Diffusion: Cell size is influenced by diffusion rates, whether for materials moving into or out of the cell or within the cell. For efficient transport, surface structures like microvilli or infoldings increase the surface area.
- Active Transport: Beyond diffusion, cells use active transport mechanisms across the plasma membrane to move macromolecules quickly and in a directed manner.
2. Cell Motility
- Forms of Movement: Cell motility includes cytoplasmic and organelle movement within cells, and external structures such as pseudopodia, lamellipodia, and flagella (e.g., sperm cells) that assist in movement or manipulation of external fluids.
- Specialized Movement Types: Cells move for functions like tissue repair (e.g., macrophages migrating through tissues) or environmental uptake (endocytosis) and secretion (exocytosis).
- Role in Muscle and Cell Division: Movement is also essential in muscle contractions and cell division processes.
3. Cell Membrane Organization
- Lipid Microdomains: Cell membranes contain lipid “rafts” (microdomains rich in sphingomyelin and cholesterol) that help segregate proteins, influencing functions like cell signaling and trafficking.
- Glycocalyx: A protective layer on cell surfaces, the glycocalyx plays a vital role in vascular permeability and cell response to mechanical stress. It’s also essential in maintaining tissue integrity and has implications in disease processes such as atherosclerosis.
4. Gene Regulation and Protein Synthesis
- RNA Interference (siRNA): Cells regulate protein synthesis through small interfering RNAs (siRNAs), which can prevent mRNA from translating into proteins. This is both a natural mechanism for regulating viral infections and a potential therapeutic approach for silencing genes involved in diseases.
5. Endoplasmic Reticulum (ER) and Golgi Apparatus
- ER-Golgi Transport: Material exchange between the ER and Golgi is essential for protein and lipid sorting. Golgins, which stabilize the Golgi structure, assist in vesicle trafficking.
- Golgi Functions: The Golgi apparatus has multiple roles: glycosylation (sugar modification), phosphorylation, and packaging of proteins and lipids into vesicles for transport. It consists of cisternae (flattened membranes) that progressively modify proteins as they move from the cis to trans faces.
- Protein Sorting: In the trans-Golgi network, proteins are sorted into vesicles based on signal sequences. These vesicles may prepare for exocytosis, or acidify their contents for lysosome transport.
6. Cell Aggregation and Communication
- Intercellular Junctions: Cells, especially epithelial cells, adhere and communicate through specialized junctions and molecular signals, forming cohesive tissue layers that organize into functional organs.
- Cell Communication: Cells send and receive signals through direct membrane interactions or molecular diffusion across intercellular spaces.
7. Size Variations
- Typical Size Range: Most cells are between 5 to 50 micrometers, e.g., lymphocytes (6 µm), red blood cells (7.5 µm), and epithelial cells (20 µm tall).
- Exceptionally Large Cells: Certain cells, like bone-resorbing osteoclasts and blood-forming megakaryocytes, can reach sizes over 200 µm. Some cells, such as neurons and muscle cells, can also extend to substantial lengths.
These structural and functional characteristics highlight the complexity of cellular architecture and underscore the specialized roles cells play in supporting life and responding to their environment.
CELLULAR ORGANIZATION
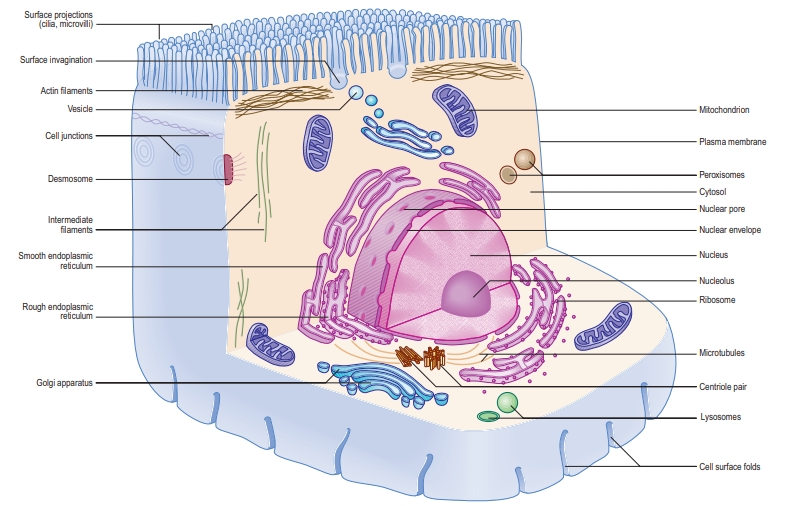
Each cell is surrounded by a plasma membrane, which encloses the cytoplasm. Most cells, except mature red blood cells, have a nucleus surrounded by a nuclear membrane. The nucleus contains the cell’s genetic material within chromosomes, as well as a nucleolus and other small structures.
Inside the cytoplasm are various membranes and organelles, which are separate compartments with specific functions. These include the rough and smooth endoplasmic reticulum, the Golgi apparatus, and vesicles. Other organelles, like lysosomes, peroxisomes, and mitochondria, each have a membrane—mitochondria and the nucleus have a double membrane, while lysosomes and peroxisomes have a single membrane.
In the cytoplasm, there are also non-membrane structures called inclusions, such as lipid droplets, glycogen clumps, and pigments like lipofuscin. Additionally, ribosomes and the cytoskeleton (a network of protein filaments) are found in the cytosol. The cytoskeleton helps maintain the cell’s shape, supports extensions like microvilli and cilia, assists in building structures like centrioles, and transports materials within the cell. The cytosol itself contains many proteins, ions, and other molecules needed for cell function.
Plasma membrane
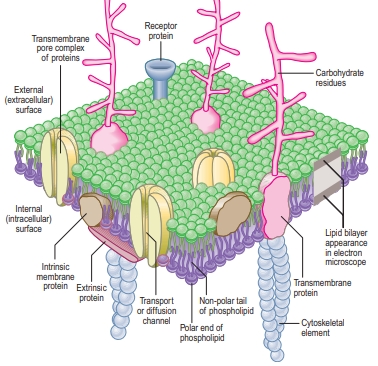
Cells are surrounded by a plasma membrane made of lipids and proteins. This flexible barrier controls what enters and exits the cell and also surrounds the nucleus. The membrane consists of a double layer of lipid molecules with water-repelling tails inside and water-attracting heads outside.
Proteins are embedded in or float within this lipid layer, allowing them to move easily. Some proteins stretch all the way across the membrane, while others attach only to the surface. These proteins help transport substances, send signals, and allow the cell to connect with other cells. Carbohydrates are linked to proteins or lipids on the outside of the membrane, helping with cell recognition and communication.
The plasma membrane is fluid, which lets proteins move and perform their functions. It creates separate areas inside the cell, keeping different processes organized. While lipid-soluble molecules like steroid hormones can pass through easily, other substances need special transport proteins to enter or leave the cell.
Additionally, the membrane helps maintain the cell’s shape and provides sites for important activities like enzyme attachment and signal reception. Inside the cell, the membrane connects to structures like the cytoskeleton, which supports cell movement and stability. Plasma membranes are made by the rough endoplasmic reticulum and the Golgi apparatus.
Cell coat (glycocalyx)
The outer surface of a cell’s plasma membrane has a unique, fuzzy layer called the glycocalyx, which is rich in carbohydrates. This layer, about 2–20 nm thick, consists of the carbohydrate parts of glycoproteins and glycolipids in the membrane.
The glycocalyx varies by cell type and contains important markers, like those that help the immune system identify cells or determine blood type in red blood cells. This layer is also important in organ transplant compatibility. In the intestine, the glycocalyx on microvilli contains enzymes for digestion, and the tightly packed microvilli increase the cell’s ability to absorb nutrients.
Cytoplasm
The cytoplasm is the gel-like substance within cells that is surrounded by the cell membrane. It consists of the cytosol—a fluid containing proteins, enzymes, and other molecules—as well as various compartments and structures that perform specific functions.
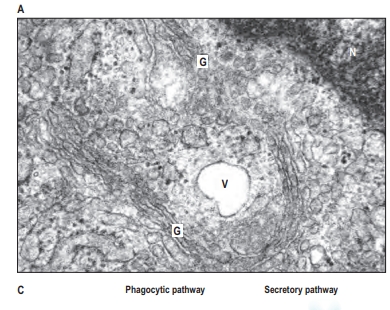
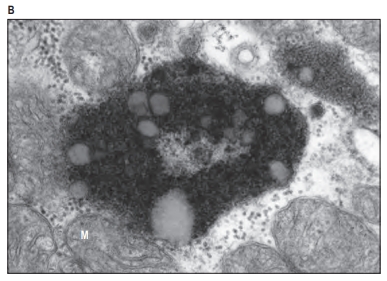
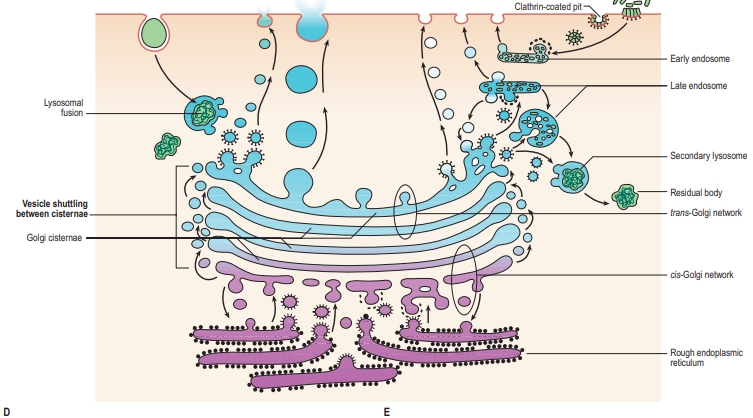
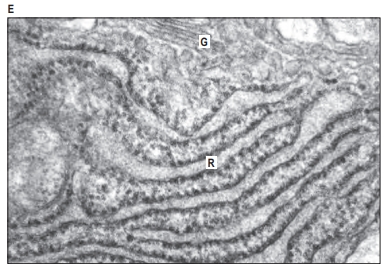
1. Cytoplasm and Its Components:
- Cytosol: The fluid part of the cytoplasm contains proteins, carbohydrates, RNA, and ribosomes, providing a medium for biochemical reactions.
- Organelles: Specialized structures, such as the endoplasmic reticulum, Golgi apparatus, lysosomes, peroxisomes, and mitochondria, each enclosed in their own membranes.
- Inclusions: Membrane-free structures like lipid droplets, glycogen particles, and pigments.
- Cytoskeleton: A network of fibers that gives the cell shape, aids in movement, and organizes the cytoplasm.
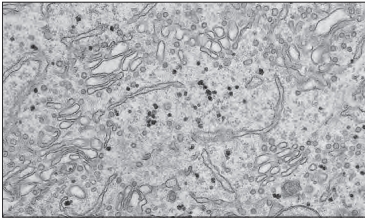
2. Endoplasmic Reticulum (ER):
- Structure: A network of membrane-lined channels that includes the rough ER (RER) with ribosomes attached and smooth ER (SER) without ribosomes.
- Functions:
- Rough ER synthesizes and folds proteins for secretion or use in membranes. Cells that produce a lot of proteins have an extensive RER.
- Smooth ER is involved in lipid and steroid production, calcium storage, and detoxification. It’s common in muscle cells and liver cells.
3. Golgi Apparatus:
- Located near the nucleus, the Golgi apparatus modifies, sorts, and packages proteins and lipids for transport.
- Vesicles from the ER bring materials to the Golgi, where they are processed and sent to various cell parts or exported outside.
4. Ribosomes and Protein Synthesis:
- Ribosomes are the protein-making machinery of the cell, found either floating in the cytosol or attached to the RER.
- Polyribosomes (groups of ribosomes) work together to translate messenger RNA (mRNA) into proteins.
5. Transport and Cell Communication:
- Vesicles coated with specific proteins help move materials between the ER, Golgi, and other areas.
- Cells in the immune system use vesicles to display antigens on their surface, allowing the immune system to recognize and respond to foreign substances.
Each structure in the cytoplasm contributes to the cell’s overall function by producing and processing materials, facilitating chemical reactions, and supporting cell structure and movement.
Exocytic (secretory) pathway
The exocytic, or secretory, pathway is the process by which cells produce and release various molecules, such as proteins and lipids, outside of the cell. This pathway is essential for communication between cells, nutrient transport, and various cellular functions. The exocytic pathway mainly involves the endoplasmic reticulum (ER), the Golgi apparatus, vesicles, and the cell membrane. Here’s a step-by-step breakdown:
1. Synthesis in the Rough Endoplasmic Reticulum (ER)
- Location and Function: The rough ER, named for its ribosome-studded surface, is the starting point of the exocytic pathway. Ribosomes on the ER’s surface synthesize proteins directly into the ER lumen.
- Protein Folding and Initial Modifications: Once inside the ER, proteins are folded into their functional structures, often with the assistance of chaperone proteins. Some initial modifications, like glycosylation (attachment of sugar molecules), may occur here to prepare proteins for their final destinations.
2. Vesicle Transport from ER to Golgi Apparatus
- Vesicle Formation: The newly synthesized and modified proteins are packaged into vesicles, small membrane-bound sacs that bud off from the ER.
- Transport to the Golgi: These vesicles move to the Golgi apparatus along microtubules, which are part of the cytoskeleton and act as “tracks” for vesicle transport.
3. Processing in the Golgi Apparatus
- Arrival at the Cis-Golgi: Vesicles fuse with the cis face (the receiving side) of the Golgi apparatus.
- Protein Modifications: Within the Golgi, proteins undergo further modifications. This includes additional glycosylation, where sugars are added or removed to modify the protein’s function or destination.
- Sorting and Packaging: As proteins move through the Golgi stacks from cis to trans (the shipping side), they are sorted and packaged based on their final destinations, which could be the plasma membrane, lysosomes, or extracellular space.
4. Vesicle Transport from the Golgi to the Cell Surface
- Formation of Secretory Vesicles: Once proteins are processed in the Golgi, they are packaged into secretory vesicles, which bud off from the trans-Golgi network.
- Types of Secretory Pathways:
- Constitutive Secretion: In this pathway, vesicles continuously transport molecules to the cell surface without any specific signal. It’s used for routine delivery of membrane proteins and lipids, as well as the release of extracellular matrix components.
- Regulated Secretion: In regulated pathways, vesicles are stored within the cell until they receive a specific signal to release their contents. This is typical in specialized cells, such as neurons or endocrine cells, where molecules like hormones or neurotransmitters are released only in response to certain stimuli.
5. Exocytosis: Fusion of Vesicles with the Plasma Membrane
- Docking and Fusion: When a vesicle reaches the cell membrane, it docks and fuses with the membrane. This fusion is facilitated by proteins called SNAREs, which help the vesicle membrane merge with the cell’s plasma membrane.
- Release of Contents: The contents of the vesicle are released outside the cell (into the extracellular space), a process known as exocytosis. For regulated secretion, this step is triggered by specific signals, such as an influx of calcium ions in neurons.
- Integration into the Membrane: Any membrane proteins or lipids within the vesicle become part of the plasma membrane, which allows cells to continuously renew their membrane composition.
Functions and Importance of the Exocytic Pathway
The exocytic pathway is crucial for:
- Cell Communication: Hormones, neurotransmitters, and cytokines released by cells enable communication between cells, which is essential for coordinating bodily functions.
- Nutrient and Waste Transport: By secreting digestive enzymes or absorbing nutrients, cells regulate nutrient transport and waste removal.
- Immune Response: Cells release antibodies and other immune molecules to detect and neutralize pathogens.
- Membrane Maintenance: By adding lipids and proteins to the membrane, exocytosis helps cells maintain and modify their surface composition, which is vital for cell signaling, adhesion, and structural integrity.
Endocytic (internalization) pathway
The endocytic, or internalization, pathway is the process by which cells take in molecules from the extracellular environment. This pathway allows cells to ingest nutrients, regulate surface receptors, and clear away debris, among other functions. The endocytic pathway is primarily mediated by structures such as the plasma membrane, vesicles, endosomes, and lysosomes. Here’s a step-by-step breakdown:
1. Initiation at the Plasma Membrane
- Recognition and Binding: The process begins when specific molecules, or ligands, outside the cell bind to receptors on the cell’s plasma membrane. This binding usually triggers the formation of an endocytic vesicle.
- Types of Endocytosis:
- Phagocytosis: Also called “cell eating,” phagocytosis is used by specialized cells (like macrophages) to engulf large particles or pathogens. This process creates large vesicles known as phagosomes.
- Pinocytosis: Known as “cell drinking,” pinocytosis involves the nonspecific uptake of extracellular fluid and dissolved substances. Small vesicles form, taking in a sampling of the surrounding fluid.
- Receptor-Mediated Endocytosis: This type is highly specific, as receptors on the cell surface bind to particular molecules (like hormones, vitamins, or antibodies). Once enough receptors are bound, the cell membrane invaginates to form a vesicle, which allows cells to efficiently internalize specific molecules even at low concentrations.
2. Vesicle Formation and Internalization
- Vesicle Formation: After binding to ligands, the plasma membrane forms a pocket that engulfs the targeted molecules, eventually pinching off to form a vesicle inside the cell. In receptor-mediated endocytosis, this process often involves clathrin, a protein that helps shape the vesicle.
- Endocytosis Vesicle: The newly formed vesicle detaches from the membrane and moves into the cytoplasm. This vesicle now contains the extracellular material along with any membrane-bound receptors involved in binding the molecules.
3. Early Endosome
- Fusion with Early Endosome: The endocytic vesicle fuses with an early endosome, an organelle that acts as a sorting station. The slightly acidic environment in the early endosome helps separate the ligands from their receptors.
- Sorting and Recycling:
- Recycling of Receptors: In some cases, the receptors that initially bound the molecules are recycled back to the cell membrane. This allows the receptors to continue capturing more molecules.
- Transition to Late Endosome: Other molecules are retained within the endosome as it matures into a late endosome, which moves deeper into the cell for further processing.
4. Late Endosome and Lysosome Fusion
- Formation of the Late Endosome: The early endosome gradually matures into a late endosome, characterized by an increasingly acidic environment.
- Fusion with Lysosomes: The late endosome fuses with a lysosome, an organelle containing hydrolytic enzymes that can break down a wide range of biomolecules.
- Degradation: Within the lysosome, the internalized molecules are broken down into their basic components (amino acids, lipids, sugars), which can then be recycled by the cell.
Functions and Importance of the Endocytic Pathway
The endocytic pathway serves several critical cellular functions:
- Nutrient Uptake: Cells use endocytosis to take in essential nutrients like cholesterol, iron, and vitamins, which are often bound to specific receptors.
- Regulation of Surface Receptors: Endocytosis allows cells to control the number and types of receptors on their surface, affecting how they respond to hormones, growth factors, and other signals.
- Defense Against Pathogens: Through phagocytosis, immune cells (like macrophages and neutrophils) can engulf and destroy pathogens, helping to protect the body from infection.
- Clearance of Cellular Debris: Cells use endocytosis to remove damaged or unnecessary material, including parts of their own cellular machinery, which helps maintain cellular health.
- Signal Regulation: Cells internalize receptors and their ligands (such as hormones) to regulate how long signals last within the cell, influencing cellular responses.
Lysosomes
Lysosomes are membrane-bound organelles found in eukaryotic cells, especially abundant in animal cells. They act as the cell’s “digestive system,” breaking down waste materials, cellular debris, and foreign substances, like pathogens, that enter the cell. Lysosomes contain powerful hydrolytic enzymes capable of degrading proteins, lipids, nucleic acids, and carbohydrates, making them essential for maintaining cellular health and homeostasis.
Structure and Composition of Lysosomes
- Membrane-Bound: Lysosomes are enclosed by a single lipid bilayer membrane, which isolates their acidic environment from the rest of the cell to prevent accidental digestion of cellular components.
- Hydrolytic Enzymes: Lysosomes contain over 50 different enzymes, including proteases, lipases, nucleases, and glycosidases. These enzymes are synthesized in the rough endoplasmic reticulum (ER), processed in the Golgi apparatus, and tagged for transport to the lysosome.
- Acidic Environment: The interior of a lysosome is highly acidic (pH around 4.5–5), which optimizes the activity of its enzymes. Proton pumps in the lysosomal membrane actively transport hydrogen ions (H+) into the organelle to maintain this acidic environment.
Formation of Lysosomes
Lysosomes are formed through the cooperation of the endoplasmic reticulum (ER) and the Golgi apparatus:
- Synthesis of Enzymes: Hydrolytic enzymes are synthesized in the ER and transported to the Golgi apparatus.
- Modification and Tagging: In the Golgi, these enzymes are modified and tagged with mannose-6-phosphate (M6P), which directs them to the lysosome.
- Packaging into Vesicles: The enzymes are packaged into vesicles, which bud off from the Golgi and eventually fuse with existing lysosomes or form new lysosomes.
Functions of Lysosomes
Lysosomes are central to several important cellular functions:
- Degradation of Waste and Cellular Debris: Lysosomes digest cellular debris, such as damaged organelles and non-functional proteins, breaking them down into their component parts for recycling.
- Autophagy: In autophagy, lysosomes help the cell survive under stress by breaking down damaged or unnecessary components. The cell encapsulates parts of itself in a double-membrane structure called an autophagosome, which fuses with a lysosome to degrade and recycle its contents.
- Pathogen Defense: In immune cells, lysosomes play a key role in fighting infection by degrading pathogens. For example, phagocytes, such as macrophages, engulf foreign substances (like bacteria) into a vesicle called a phagosome, which then fuses with a lysosome to form a phagolysosome. Here, the pathogens are destroyed by lysosomal enzymes.
- Cellular Recycling: After digestion, lysosomes release monomers like amino acids, fatty acids, and sugars back into the cytoplasm, where they are reused by the cell for energy or to build new structures.
- Programmed Cell Death (Apoptosis): In some cases, lysosomes participate in cell death by releasing their enzymes into the cytoplasm, initiating the process of cellular breakdown. This is a controlled process essential for development and cellular turnover in multicellular organisms.
Lysosomal Storage Diseases
Lysosomal storage diseases are a group of genetic disorders caused by mutations that affect lysosomal enzyme function. Without the necessary enzymes, certain substrates accumulate within lysosomes, leading to cell damage. Examples include:
- Tay-Sachs Disease: Caused by a deficiency in the enzyme hexosaminidase A, leading to the buildup of a fatty substance in the brain and nerve cells.
- Gaucher Disease: Caused by a deficiency in the enzyme glucocerebrosidase, resulting in the accumulation of glucocerebroside, particularly affecting the liver, spleen, and bone marrow.
- Pompe Disease: Involves the buildup of glycogen in cells due to a deficiency of the enzyme acid alpha-glucosidase.
Lysosomal dysfunction
Lysosomal dysfunction refers to the impairment of lysosomal function, which can disrupt cellular waste breakdown, recycling, and other essential lysosomal activities. This dysfunction can lead to the accumulation of undigested molecules within cells, eventually causing cellular damage and disease. Lysosomal dysfunction can arise from genetic mutations, environmental factors, or age-related changes and has been linked to various lysosomal storage diseases, neurodegenerative disorders, and other health issues.
Causes of Lysosomal Dysfunction
- Genetic Mutations: Many lysosomal enzymes and structural proteins are encoded by specific genes, and mutations in these genes can impair enzyme function or lysosome integrity. Such mutations often result in lysosomal storage diseases (LSDs), where the lack of one or more enzymes prevents the breakdown of certain substrates, leading to their accumulation.
- Environmental and Chemical Factors: Some environmental toxins, heavy metals, and pollutants can impair lysosomal function by either directly damaging lysosomal membranes or altering enzyme activity within the lysosome.
- Age-Related Decline: As cells age, lysosomal function generally declines. This is due to factors like reduced enzyme efficiency, accumulation of cellular waste, and oxidative damage. This age-related dysfunction contributes to the progression of age-associated diseases.
- Cellular Stress: Oxidative stress, nutrient deprivation, and other cellular stresses can compromise lysosomal membrane stability and enzyme function, affecting the lysosome’s ability to perform autophagy and other critical processes.
Consequences of Lysosomal Dysfunction
- Lysosomal Storage Diseases (LSDs): Lysosomal storage diseases are a group of over 50 rare genetic disorders caused by defects in lysosomal enzymes. This causes the accumulation of specific substrates within lysosomes, leading to cell damage and symptoms that vary depending on the disease type and the tissues affected. Examples include:
- Gaucher Disease: Accumulation of glucocerebroside primarily in the liver, spleen, and bone marrow.
- Tay-Sachs Disease: Accumulation of GM2 gangliosides in neurons, leading to neurodegeneration.
- Fabry Disease: Build-up of globotriaosylceramide, affecting blood vessels, kidneys, and the heart.
- Neurodegenerative Diseases: Lysosomal dysfunction is increasingly linked to neurodegenerative diseases like Alzheimer’s, Parkinson’s, and Huntington’s. In these diseases, lysosomal impairment contributes to the buildup of toxic protein aggregates that damage neurons:
- Alzheimer’s Disease: Defective lysosomal autophagy can lead to the accumulation of amyloid-beta plaques and tau protein tangles.
- Parkinson’s Disease: Mutations in genes like GBA (encoding glucocerebrosidase) are associated with lysosomal dysfunction and contribute to alpha-synuclein accumulation, a key factor in Parkinson’s pathology.
- Impaired Autophagy: Autophagy is the process by which cells degrade and recycle damaged organelles and proteins. Lysosomes play a crucial role in autophagy by digesting the contents of autophagosomes. Dysfunctional lysosomes disrupt autophagy, leading to the accumulation of damaged components, reduced energy supply, and increased cell stress.
- Cellular Toxicity and Inflammation: Accumulated waste products in lysosomes can become toxic, damaging other cellular components. Lysosomal dysfunction can also lead to the release of partially digested molecules, triggering inflammatory responses. In immune cells, defective lysosomes can impair pathogen clearance, making the body more susceptible to infections.
- Tumorigenesis: Lysosomal dysfunction has been implicated in cancer development and progression. Impaired lysosomal degradation can disrupt normal cell death pathways and autophagy, promoting uncontrolled cell growth and survival. Some cancer cells even adapt to survive under low-nutrient conditions by altering their lysosomal function to maintain energy levels.
Potential Treatments for Lysosomal Dysfunction
- Enzyme Replacement Therapy (ERT): For some lysosomal storage diseases, enzyme replacement therapy provides patients with functional versions of the missing or defective enzyme. This helps break down the accumulated substrates and alleviate symptoms. ERT is available for diseases like Gaucher, Fabry, and Pompe.
- Substrate Reduction Therapy (SRT): SRT aims to reduce the production of the substrates that accumulate due to lysosomal enzyme deficiencies. By lowering substrate levels, SRT minimizes the burden on dysfunctional lysosomes and mitigates the accumulation of toxic materials.
- Gene Therapy: Gene therapy attempts to correct genetic mutations responsible for lysosomal dysfunction. By introducing functional copies of defective genes into cells, gene therapy could restore proper enzyme function in lysosomal storage diseases.
- Autophagy Inducers: In neurodegenerative and age-related diseases, compounds that stimulate autophagy might help reduce the buildup of damaged proteins and cellular components, supporting lysosomal function and cellular health.
- Lysosome-Targeted Drugs: Some treatments focus on stabilizing lysosomal membranes, enhancing enzyme function, or regulating lysosomal pH to improve overall function. This area of research is expanding, particularly in targeting age-related lysosomal decline.
26S proteasome
The 26S proteasome is a large protein complex found in eukaryotic cells that plays a central role in protein degradation and cellular regulation. It is part of the ubiquitin-proteasome system (UPS), which tags unwanted or damaged proteins with a small protein called ubiquitin, marking them for degradation by the proteasome. This system is essential for controlling the quality, quantity, and function of proteins within cells and helps regulate processes like cell cycle progression, signal transduction, and responses to cellular stress.
Structure of the 26S Proteasome
The 26S proteasome is a multi-subunit complex, made up of two main parts:
- 20S Core Particle (CP):
- The 20S core particle is cylindrical and consists of four stacked rings: two outer alpha rings and two inner beta rings, each made up of seven subunits.
- The alpha rings control the entry of proteins into the core, while the beta rings contain the active proteolytic (protein-degrading) sites.
- The 20S core particle is responsible for the actual degradation of proteins, breaking them down into small peptides.
- 19S Regulatory Particle (RP):
- The 19S regulatory particle attaches to either one or both ends of the 20S core particle to form the 26S proteasome complex.
- It is divided into two subcomplexes: the “base” and the “lid.”
- Base: The base contains ATPase subunits that unfold the protein substrates and translocate them into the core particle for degradation.
- Lid: The lid recognizes and removes ubiquitin tags from the substrate before it enters the core particle.
- The 19S regulatory particle plays a crucial role in recognizing, binding, and preparing ubiquitinated proteins for degradation.
Function of the 26S Proteasome
The primary function of the 26S proteasome is to degrade polyubiquitinated proteins into small peptides. This process occurs in several key steps:
- Recognition and Binding:
- Proteins targeted for degradation are first tagged with chains of ubiquitin through the action of three enzymes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase).
- The 19S regulatory particle of the proteasome recognizes and binds to these ubiquitin-tagged proteins.
- Ubiquitin Removal and Unfolding:
- Before the tagged protein enters the 20S core particle, the 19S lid removes the ubiquitin chain, which is recycled for future tagging processes.
- ATPase enzymes in the 19S base then unfold the protein, allowing it to pass into the narrow channel of the 20S core for degradation.
- Protein Degradation:
- The unfolded protein enters the 20S core particle, where it is broken down by proteolytic enzymes in the beta rings.
- The proteasome cleaves the protein into short peptide fragments, typically 3-25 amino acids in length.
- Peptide and Amino Acid Recycling:
- The degraded peptides are then released into the cytoplasm, where they can be further broken down into individual amino acids and recycled to create new proteins.
Functions and Importance of the 26S Proteasome
The 26S proteasome plays several essential roles in cellular homeostasis and regulation:
- Protein Quality Control: By degrading misfolded or damaged proteins, the proteasome prevents the accumulation of potentially toxic proteins that could disrupt cellular function.
- Regulation of Cellular Processes: The proteasome regulates key processes like the cell cycle, transcription, and signal transduction by controlling the levels of specific proteins. For example, the proteasome degrades cyclins and other regulatory proteins, allowing cells to progress through different phases of the cell cycle.
- Response to Stress: In conditions of stress, such as oxidative stress or nutrient deprivation, the proteasome helps degrade damaged proteins, supporting cellular recovery and survival.
- Antigen Presentation: In immune cells, the proteasome generates peptides from degraded proteins, which are then presented on the cell surface in complex with MHC (major histocompatibility complex) molecules. This process is critical for the immune system’s ability to recognize and respond to pathogens.
Clinical Significance of the 26S Proteasome
Dysfunction in the ubiquitin-proteasome system is associated with several diseases:
- Neurodegenerative Diseases: Disorders like Alzheimer’s, Parkinson’s, and Huntington’s disease involve the accumulation of abnormal proteins due to impaired proteasome function. These aggregated proteins disrupt cellular processes and lead to neuronal damage.
- Cancer: Cancer cells often have altered proteasome activity, which can allow them to evade normal regulatory mechanisms that would otherwise control cell proliferation and survival. Proteasome inhibitors, like bortezomib, are used as a treatment in certain cancers (e.g., multiple myeloma) to induce apoptosis in cancer cells by disrupting their protein homeostasis.
- Autoimmune Diseases: Malfunctioning proteasomes can impact immune response, leading to autoimmune conditions where the immune system mistakenly targets the body’s own cells.
Peroxisomes
Peroxisomes are small, membrane-bound organelles found in almost all eukaryotic cells. They play a crucial role in various metabolic processes, primarily involved in lipid metabolism and detoxification of reactive oxygen species (ROS). Peroxisomes contain enzymes that catalyze oxidative reactions, producing hydrogen peroxide (H₂O₂) as a byproduct, which is then broken down by the enzyme catalase to prevent cellular damage.
Structure of Peroxisomes
- Membrane-Bound: Peroxisomes are enclosed by a single lipid bilayer membrane that separates their contents from the rest of the cell’s cytoplasm.
- Enzyme-Rich Matrix: Inside the peroxisome, there is a dense matrix containing enzymes necessary for its functions, such as oxidases and catalase.
- Dynamic Organelles: Peroxisomes can grow and divide independently, adapting their numbers and enzyme content based on the cell’s needs, which is especially important in liver, kidney, and brain cells that are heavily involved in detoxification and metabolism.
Functions of Peroxisomes
Peroxisomes are essential for cellular homeostasis and carry out several key functions:
- Lipid Metabolism:
- Fatty Acid β-Oxidation: Peroxisomes are responsible for the β-oxidation of long-chain and very-long-chain fatty acids, breaking them down into shorter fatty acids and acetyl-CoA molecules. While mitochondria also perform β-oxidation, peroxisomes specialize in handling longer fatty acid chains that cannot be directly processed by mitochondria.
- Synthesis of Plasmalogens: Peroxisomes are involved in synthesizing plasmalogens, which are specialized lipids essential for the integrity and function of cell membranes, particularly in nerve cells and heart tissue.
- Detoxification:
- Decomposition of Hydrogen Peroxide: Many reactions in peroxisomes produce hydrogen peroxide (H₂O₂), a potentially harmful byproduct. The enzyme catalase within peroxisomes converts H₂O₂ into water and oxygen, preventing oxidative damage to cellular components.
- Metabolism of Reactive Oxygen Species (ROS): Peroxisomes help detoxify various ROS, protecting cells from oxidative stress and damage.
- Amino Acid and Polyamine Metabolism: Peroxisomes play a role in the metabolism of certain amino acids and polyamines, such as lysine, glycine, and polyamines involved in cellular growth and signaling.
- Detoxification of Harmful Substances: In the liver and kidneys, peroxisomes break down toxins, drugs, and other harmful substances. They contain enzymes that oxidize these molecules, making them less harmful and easier to excrete.
- Cholesterol and Bile Acid Synthesis: Peroxisomes participate in the early steps of cholesterol synthesis and in the conversion of cholesterol into bile acids, which are critical for digesting and absorbing dietary fats.
- Regulation of Cellular Signaling: Peroxisomes are involved in signaling pathways that regulate inflammation and oxidative stress responses. They interact with mitochondria to coordinate cellular responses to metabolic changes and stress.
Peroxisomal Biogenesis and Enzyme Import
Peroxisomes are unique in their ability to import enzymes directly from the cytoplasm:
- Peroxisomal Targeting Signal (PTS): Enzymes destined for peroxisomes contain specific amino acid sequences known as peroxisomal targeting signals (PTS) that guide them to the peroxisome.
- Peroxins (PEX Proteins): A family of proteins called peroxins (encoded by PEX genes) assists in recognizing and transporting enzymes into peroxisomes. Defects in these genes lead to peroxisomal biogenesis disorders.
Peroxisomes can form de novo by budding off from the endoplasmic reticulum or by growing and dividing from existing peroxisomes. This ability to self-replicate is essential for maintaining peroxisome numbers in rapidly dividing cells and tissues with high metabolic demand.
Peroxisomal Disorders
Genetic mutations that affect peroxisome biogenesis or enzyme function can lead to peroxisomal disorders. Some examples include:
- Zellweger Spectrum Disorders (ZSD): ZSDs are a group of genetic conditions caused by mutations in PEX genes, resulting in defective peroxisome biogenesis. This leads to a range of severe symptoms, including developmental delays, neurological impairment, and abnormalities in liver, kidney, and bone function.
- Adrenoleukodystrophy (ALD): ALD is a disorder that affects the breakdown of very-long-chain fatty acids, primarily impacting the adrenal glands and nervous system. This results in the buildup of fatty acids that damage myelin, the protective covering around nerve cells.
- Refsum Disease: Refsum disease is caused by defects in the breakdown of phytanic acid, a type of fatty acid found in certain foods. This leads to the accumulation of phytanic acid, causing neurological and visual impairments.
Interactions with Mitochondria and Other Organelles
Peroxisomes interact closely with mitochondria and other organelles to maintain cellular homeostasis:
- Shared Metabolic Pathways: Peroxisomes and mitochondria collaborate in fatty acid oxidation, ROS metabolism, and cellular energy regulation.
- Reactive Oxygen Species (ROS) Signaling: Both peroxisomes and mitochondria produce ROS as byproducts. Through coordinated signaling, they balance ROS levels to prevent oxidative stress and regulate cell survival.
- Lipid Exchange: Peroxisomes work with the endoplasmic reticulum (ER) to synthesize and transfer lipids, including cholesterol and plasmalogens, which are essential for cell membrane structure and function.
Mitochondria
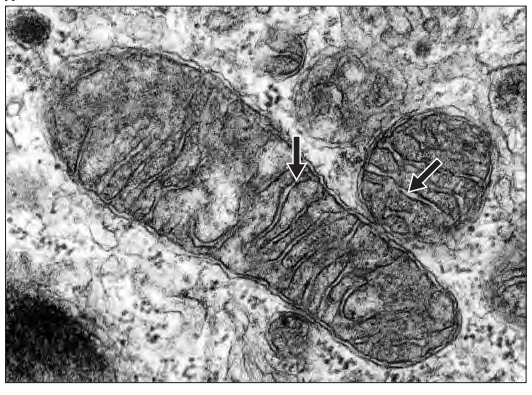
Mitochondria are membrane-bound organelles found in nearly all eukaryotic cells, often referred to as the “powerhouses” of the cell due to their role in generating energy in the form of adenosine triphosphate (ATP). They are unique among organelles because they contain their own DNA (mtDNA) and are thought to have originated from an ancient symbiotic relationship between primitive eukaryotic cells and a type of bacteria.
Structure of Mitochondria
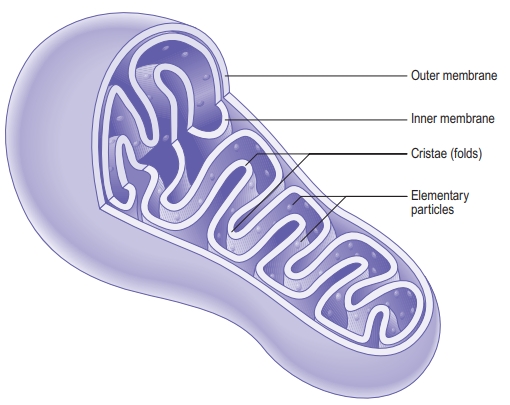
Mitochondria have a double-membrane structure that allows for compartmentalized reactions:
- Outer Membrane:
- The outer membrane is smooth and permeable to small molecules and ions due to porin proteins that form channels. It allows ions and small molecules to enter and leave the mitochondrion freely.
- Intermembrane Space:
- The intermembrane space lies between the outer and inner membranes. This space is important for the electron transport chain and ATP synthesis.
- Inner Membrane:
- The inner membrane is highly folded into structures called cristae, which increase the surface area for ATP production. It is impermeable to most molecules, creating a specialized environment for the mitochondrion’s inner processes.
- Embedded within the inner membrane are proteins and enzymes essential for the electron transport chain and ATP production.
- Matrix:
- The matrix is the innermost compartment of the mitochondrion, containing mitochondrial DNA, ribosomes, and enzymes that facilitate the citric acid cycle (Krebs cycle) and fatty acid oxidation.
Functions of Mitochondria
Mitochondria are essential for energy production and other key cellular processes:
- ATP Production (Cellular Respiration):
- Mitochondria are the primary site for ATP production through a process called cellular respiration, which involves three main stages:
- Glycolysis: Occurs in the cell’s cytoplasm, where glucose is broken down into pyruvate, producing a small amount of ATP.
- Citric Acid Cycle (Krebs Cycle): Takes place in the mitochondrial matrix. Acetyl-CoA, derived from pyruvate and fatty acids, enters the cycle and produces high-energy electron carriers NADH and FADH₂.
- Electron Transport Chain (ETC) and Oxidative Phosphorylation: Occur in the inner membrane. NADH and FADH₂ donate electrons to the ETC, which creates a proton gradient across the inner membrane. ATP synthase then uses this gradient to produce ATP as protons flow back into the matrix.
- Mitochondria are the primary site for ATP production through a process called cellular respiration, which involves three main stages:
- Calcium Storage and Regulation:
- Mitochondria store calcium ions and regulate their levels within the cell, which is important for cell signaling, muscle contraction, and neurotransmitter release.
- Apoptosis (Programmed Cell Death):
- Mitochondria play a key role in apoptosis by releasing pro-apoptotic factors like cytochrome c into the cytoplasm. This triggers a cascade of events that lead to controlled cell death, which is essential for development and cellular turnover.
- Metabolism of Reactive Oxygen Species (ROS):
- During ATP production, mitochondria produce reactive oxygen species as byproducts. To protect the cell, mitochondria contain enzymes like superoxide dismutase (SOD) and catalase that neutralize ROS, preventing cellular damage.
- Steroid and Heme Synthesis:
- Mitochondria contribute to the synthesis of steroids, cholesterol, and heme (a component of hemoglobin) through various biochemical pathways.
Mitochondrial DNA and Inheritance
- Mitochondrial DNA (mtDNA): Mitochondria have their own circular DNA, which encodes a small number of proteins essential for mitochondrial function, as well as tRNAs and rRNAs for mitochondrial protein synthesis. However, most mitochondrial proteins are encoded by nuclear DNA and imported into mitochondria.
- Maternal Inheritance: Mitochondrial DNA is inherited exclusively from the mother, as mitochondria from the sperm are typically not retained during fertilization. This unique inheritance pattern allows scientists to trace maternal lineage through mtDNA analysis.
Mitochondrial Biogenesis and Dynamics
Mitochondria can grow and divide independently of the cell cycle:
- Fission and Fusion: Mitochondria are dynamic organelles that undergo fission (splitting) and fusion (joining together) to regulate their number, shape, and function. These processes allow cells to adapt to metabolic demands, repair damaged mitochondria, and ensure even distribution during cell division.
- Biogenesis: Mitochondrial biogenesis is the process by which new mitochondria are produced. This process is regulated by signaling pathways that respond to cellular energy needs, especially during increased energy demand such as exercise or growth.
Mitochondrial Dysfunction and Disease
Dysfunction in mitochondria is associated with a range of diseases:
- Mitochondrial Disorders: Mutations in mtDNA or nuclear genes encoding mitochondrial proteins can lead to mitochondrial diseases, which often affect tissues with high energy demands, like muscles, brain, and heart. Examples include:
- Leigh Syndrome: A severe neurological disorder caused by defects in mitochondrial function.
- Mitochondrial Myopathy: A group of diseases affecting muscle tissue, often causing weakness and fatigue.
- Neurodegenerative Diseases: Mitochondrial dysfunction contributes to neurodegenerative diseases like Alzheimer’s, Parkinson’s, and Huntington’s disease. Damage to mitochondrial DNA or oxidative stress from excess ROS can lead to neuronal cell death, impacting brain function.
- Aging: Mitochondrial dysfunction has been implicated in the aging process. Over time, mitochondrial DNA accumulates mutations, and the cell’s ability to neutralize ROS diminishes, leading to cellular damage and reduced energy production, which are linked to aging and age-related diseases.
- Metabolic Disorders: Since mitochondria are central to energy metabolism, defects in mitochondrial function can lead to metabolic disorders like diabetes, obesity, and insulin resistance.
- Cancer: Mitochondrial dysfunction can contribute to cancer by altering cell metabolism, supporting uncontrolled growth, and allowing cancer cells to survive in low-oxygen environments.
Cell signalling
Cell signaling, also known as cell communication, refers to the complex system of interactions and mechanisms that cells use to communicate with one another and respond to their environment. Through cell signaling, cells are able to coordinate actions, maintain homeostasis, and adapt to changes. The process involves signaling molecules, receptors, pathways, and cascades that transmit information from one cell to another or within the same cell.
Key Components of Cell Signaling
- Signaling Molecules (Ligands): These are molecules that act as signals to initiate the cell signaling process. They can include:
- Hormones: Chemical messengers secreted into the bloodstream, affecting distant target cells (e.g., insulin, estrogen).
- Neurotransmitters: Chemicals released by neurons to transmit signals across synapses to other neurons or muscle cells (e.g., dopamine, serotonin).
- Cytokines: Signaling proteins involved in immune responses (e.g., interleukins, interferons).
- Growth Factors: Molecules that stimulate cell growth, proliferation, and differentiation (e.g., epidermal growth factor).
- Local Mediators: Molecules that affect nearby cells, acting in a paracrine manner (e.g., nitric oxide, prostaglandins).
- Receptors: Receptors are proteins located on the cell surface or within cells that specifically bind to signaling molecules, initiating the signaling cascade. Types of receptors include:
- Cell Surface Receptors: Located on the plasma membrane, they bind to hydrophilic ligands that cannot cross the membrane (e.g., G-protein-coupled receptors, receptor tyrosine kinases).
- Intracellular Receptors: Located within the cytoplasm or nucleus, they bind to hydrophobic ligands that can cross the cell membrane (e.g., steroid hormone receptors).
- Signal Transduction Pathways: Signal transduction refers to the process by which a signal on a cell’s surface is converted into a specific cellular response. It involves multiple molecules and steps, including:
- Secondary Messengers: Small molecules that relay signals received by receptors to target molecules inside the cell (e.g., cAMP, calcium ions).
- Protein Kinases and Phosphatases: Enzymes that modify other proteins by adding or removing phosphate groups, thus altering their activity.
- Transcription Factors: Proteins that regulate gene expression in response to signaling pathways, affecting cell behavior over longer timescales.
- Cellular Responses: The final effect of signaling, where cells respond by altering their activity, behavior, or gene expression, resulting in outcomes like growth, differentiation, survival, apoptosis, or movement.
Types of Cell Signaling
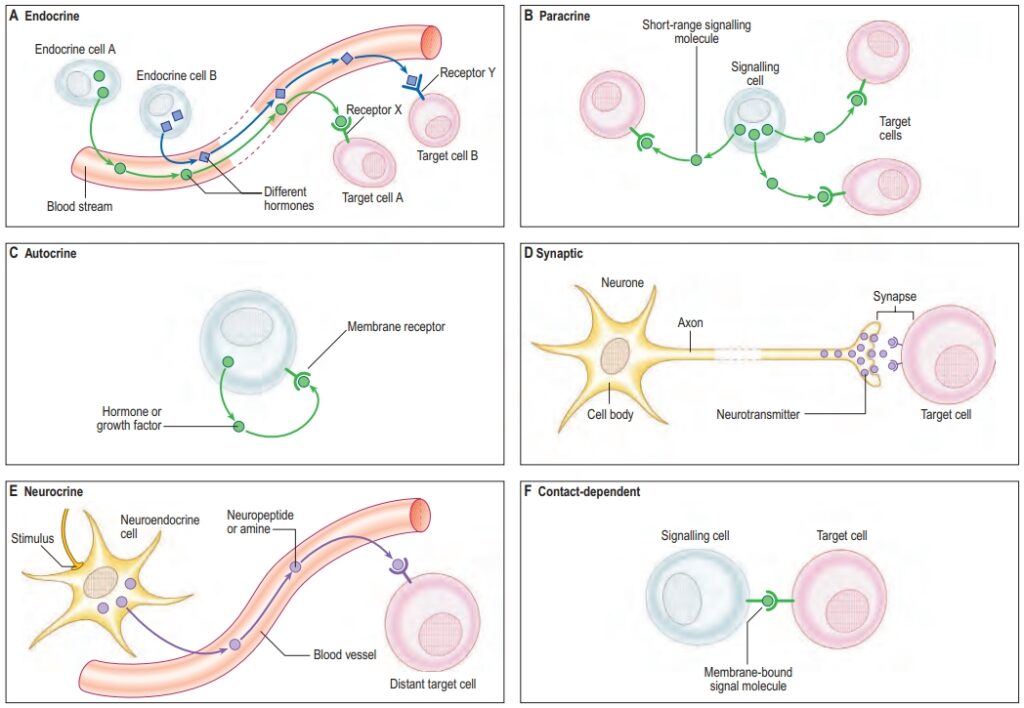
Cell signaling can be classified based on the distance between the signaling cell and the target cell:
- Autocrine Signaling: The cell produces and responds to its own signals. This type of signaling is important for self-regulation and is often seen in immune cells and during cell differentiation.
- Paracrine Signaling: The signaling molecule acts on nearby cells. Paracrine signaling is common in tissue communication, immune responses, and wound healing.
- Endocrine Signaling: Signaling molecules, such as hormones, travel through the bloodstream to distant target cells. Endocrine signaling allows for coordination across different parts of the body (e.g., insulin regulates glucose metabolism).
- Synaptic Signaling: Specialized form of paracrine signaling in which neurotransmitters are released from neurons across a synapse to target neurons or muscle cells. Synaptic signaling is essential for the nervous system’s function.
- Juxtacrine Signaling: Also known as contact-dependent signaling, it occurs when signaling molecules remain attached to the surface of the signaling cell and interact with receptors on adjacent cells, important in cell adhesion and tissue development.
Major Cell Signaling Pathways
Several key pathways are fundamental to cell signaling:
- G-Protein-Coupled Receptor (GPCR) Pathway:
- GPCRs are the largest family of cell surface receptors, which bind to extracellular ligands and activate G-proteins inside the cell.
- When activated, G-proteins dissociate into subunits that can activate other signaling molecules like adenylyl cyclase, leading to the production of secondary messengers such as cAMP.
- GPCR signaling controls processes like vision, taste, and neurotransmission.
- Receptor Tyrosine Kinase (RTK) Pathway:
- RTKs are transmembrane receptors that, upon binding a ligand, dimerize and auto-phosphorylate tyrosine residues, activating downstream signaling cascades.
- The RTK pathway activates signaling cascades such as the MAPK/ERK pathway, which regulates cell proliferation, differentiation, and survival.
- Examples include growth factor receptors and insulin receptors.
- Notch Signaling Pathway:
- In juxtacrine signaling, Notch receptors on one cell interact with ligands on an adjacent cell, leading to the cleavage of Notch and its movement to the nucleus to influence gene expression.
- Notch signaling is crucial for cell fate determination during development.
- Wnt Signaling Pathway:
- The Wnt pathway is involved in regulating cell proliferation and differentiation, particularly during embryonic development.
- Binding of Wnt to its receptor, Frizzled, stabilizes β-catenin, allowing it to enter the nucleus and promote transcription of target genes.
- Nuclear Receptor Pathway:
- Intracellular receptors, such as steroid hormone receptors, bind to hydrophobic ligands that diffuse through the membrane.
- The receptor-ligand complex then moves to the nucleus and binds to specific DNA sequences, regulating gene transcription.
- This pathway regulates processes such as metabolism, immune response, and reproductive function.
- JAK-STAT Signaling Pathway:
- JAK-STAT is activated by cytokines binding to receptors associated with Janus kinases (JAKs). Activated JAKs phosphorylate STAT proteins, which then dimerize and move to the nucleus to regulate gene transcription.
- This pathway is essential for immune function, cell growth, and hematopoiesis.
- PI3K-AKT Pathway:
- Activated by growth factors, PI3K phosphorylates lipids in the plasma membrane, which then recruit AKT, a kinase that promotes cell survival and growth.
- The PI3K-AKT pathway is key in cell survival, metabolism, and apoptosis inhibition.
- NF-κB Pathway:
- NF-κB is a transcription factor that, upon activation by signals like cytokines, enters the nucleus and initiates transcription of genes involved in immune and inflammatory responses.
- NF-κB signaling is involved in inflammation, immune response, and cell survival.
Cell Signaling Cascades and Amplification
Cell signaling often involves a cascade of events, allowing for signal amplification:
- Amplification occurs when a single activated receptor initiates a cascade that activates many downstream molecules, significantly enhancing the signal’s impact.
- Cross-Talk: Different signaling pathways can interact and influence each other, allowing cells to integrate multiple signals to make coordinated responses.
Cellular Responses to Signaling
Signaling cascades culminate in a cellular response, which may include:
- Gene Expression Changes: Activation of transcription factors can lead to upregulation or downregulation of specific genes.
- Metabolic Changes: Certain signaling pathways influence enzyme activity, leading to alterations in cellular metabolism.
- Cell Growth and Proliferation: Growth factor signals can lead to cell cycle progression and proliferation.
- Cell Survival or Apoptosis: Cells can either activate survival pathways or initiate programmed cell death based on external signals.
- Differentiation: Cells can change their phenotype in response to specific developmental signals.
- Immune Responses: Cytokines and other immune signals direct immune cell activation, movement, and response to pathogens.
Aberrations in Cell Signaling and Disease
Dysregulation of cell signaling pathways is associated with numerous diseases:
- Cancer: Mutations in growth factor signaling pathways (e.g., RTK and PI3K-AKT) can lead to uncontrolled cell division.
- Autoimmune Diseases: Abnormal activation of signaling pathways like NF-κB can lead to immune dysregulation and autoimmunity.
- Metabolic Disorders: Defects in insulin signaling pathways contribute to conditions like diabetes.
- Neurodegenerative Diseases: Disruptions in neurotransmitter signaling can lead to neurodegenerative diseases like Parkinson’s and Alzheimer’s.
Cytoskeleton
The cytoskeleton is a dynamic network of protein filaments and tubules within the cell that provides structural support, maintains cell shape, enables cellular movement, and facilitates intracellular transport. It plays an essential role in a wide range of cellular processes, including cell division, organelle positioning, and cell signaling. The cytoskeleton is composed of three main types of filaments: microfilaments (actin filaments), intermediate filaments, and microtubules, each with distinct structures, functions, and associated proteins.
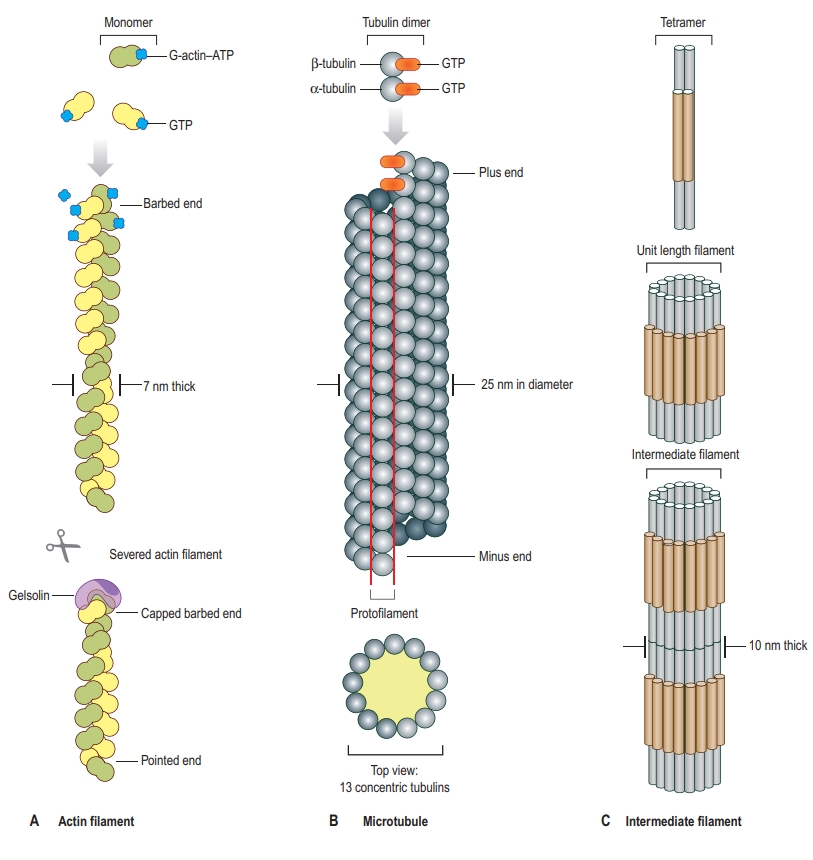
Main Components of the Cytoskeleton
- Microfilaments (Actin Filaments)
- Structure:
- Microfilaments are thin, helical filaments approximately 7 nm in diameter, primarily composed of the protein actin.
- Actin exists in two forms: globular actin (G-actin) and filamentous actin (F-actin). G-actin monomers polymerize to form F-actin chains.
- Functions:
- Cell Shape and Mechanical Support: Actin filaments provide structural support, helping maintain the cell’s shape.
- Cell Motility: Actin filaments are crucial in processes like cell crawling, where they polymerize at the leading edge of the cell to push the membrane forward.
- Muscle Contraction: In muscle cells, actin interacts with myosin to enable contraction. This interaction also facilitates cellular movement in non-muscle cells.
- Cytokinesis: During cell division, actin filaments form a contractile ring that pinches the cell into two daughter cells.
- Associated Proteins:
- Myosin: A motor protein that interacts with actin filaments to generate force and movement.
- Tropomyosin and Troponin: Regulatory proteins that control muscle contraction by modulating actin-myosin interactions.
- Cofilin and Profilin: Proteins involved in actin filament dynamics, helping to disassemble and assemble actin monomers as needed.
- Structure:
- Intermediate Filaments
- Structure:
- Intermediate filaments are rope-like fibers with a diameter of about 10 nm, providing mechanical strength to cells.
- Unlike actin filaments and microtubules, intermediate filaments do not undergo rapid assembly and disassembly; they are more stable.
- The composition of intermediate filaments varies depending on the cell type. They include different proteins like keratins, vimentin, and lamins.
- Functions:
- Structural Support: Intermediate filaments provide tensile strength, allowing cells to withstand mechanical stress.
- Cell and Tissue Integrity: In epithelial cells, keratin filaments link to desmosomes, helping cells resist stretching forces and providing tissue integrity.
- Nuclear Structure: The nuclear lamina, a network of intermediate filaments (lamins) beneath the nuclear envelope, supports nuclear shape and organizes chromatin.
- Signal Transduction: Some intermediate filaments interact with signaling proteins, influencing cellular responses to mechanical stress.
- Associated Proteins:
- Plectin: Links intermediate filaments to other cytoskeletal components, such as actin filaments and microtubules.
- Desmoplakin: Connects intermediate filaments to desmosomes in epithelial cells, providing strength to cell-cell junctions.
- Structure:
- Microtubules
- Structure:
- Microtubules are hollow tubes approximately 25 nm in diameter, composed of alpha and beta tubulin dimers that polymerize in a head-to-tail arrangement to form a hollow cylinder.
- Microtubules have polarity, with a “plus” end, where tubulin addition occurs more readily, and a “minus” end, which is typically anchored to the microtubule-organizing center (MTOC).
- Functions:
- Intracellular Transport: Microtubules act as tracks for motor proteins, such as kinesin and dynein, which transport vesicles, organelles, and other cargoes within the cell.
- Cell Division: Microtubules form the mitotic spindle, which separates chromosomes during cell division.
- Cell Shape and Support: Microtubules help maintain cell shape, especially in cells with asymmetric structures like neurons.
- Cilia and Flagella Movement: In ciliated and flagellated cells, microtubules form the core structure (axoneme) that enables movement.
- Associated Proteins:
- Kinesin and Dynein: Motor proteins that “walk” along microtubules to transport cellular cargo.
- Tau and MAPs (Microtubule-Associated Proteins): Stabilize microtubules and regulate their dynamics, crucial in neurons for maintaining axon structure.
- Structure:
Cytoskeleton-Related Cellular Processes
- Cell Motility and Migration
- Cell movement relies on actin filaments and microtubules. Actin polymerization at the cell’s leading edge creates protrusions, such as lamellipodia and filopodia, that allow the cell to crawl forward.
- The coordination of actin and microtubule dynamics, along with myosin activity, enables cells to migrate in response to external signals.
- Intracellular Transport
- Motor proteins (dynein and kinesin) move along microtubules, transporting organelles, vesicles, and proteins throughout the cell.
- Actin filaments also support short-range transport near the cell membrane and within the cell cortex.
- Cell Division
- During mitosis, the microtubule-based mitotic spindle separates duplicated chromosomes.
- Actin filaments form the contractile ring during cytokinesis, leading to cell cleavage and division into two daughter cells.
- Signal Transduction
- The cytoskeleton acts as a platform for signaling molecules, enabling the cell to respond to changes in its environment.
- Mechanical signals, such as those from the extracellular matrix, can be transmitted to the cell interior via the cytoskeleton, influencing gene expression and cell behavior.
- Endocytosis and Vesicle Transport
- Actin filaments assist in forming membrane invaginations during endocytosis.
- Microtubules guide vesicles from endocytic vesicles to other cellular locations for recycling or degradation.
- Maintenance of Cell Shape and Polarity
- The cytoskeleton maintains the shape of cells, especially those with specialized forms, such as neurons and epithelial cells.
- In polarized cells, like epithelial cells, the cytoskeleton ensures that specific proteins and organelles are localized to particular cellular regions.
Cytoskeletal Dynamics
The cytoskeleton is highly dynamic, constantly undergoing polymerization and depolymerization. This dynamism is regulated by various factors and proteins:
- Polymerization and Depolymerization:
- Actin Treadmilling: Actin filaments grow at the plus end (barbed end) and shrink at the minus end (pointed end), allowing rapid remodeling.
- Dynamic Instability of Microtubules: Microtubules undergo phases of growth and shrinkage, driven by GTP hydrolysis, allowing them to explore the cellular space rapidly.
- Regulatory Proteins:
- Actin-Binding Proteins: Proteins like profilin, cofilin, and gelsolin regulate actin filament dynamics.
- Microtubule-Associated Proteins (MAPs): Stabilize or destabilize microtubules, affecting their growth and shrinkage.
- Cross-Linking and Bundling:
- Cross-linking proteins like filamin and spectrin bundle or link filaments to create more complex structures, reinforcing the cytoskeleton and providing mechanical resilience.
Cytoskeleton and Disease
Dysfunction in the cytoskeleton is linked to various diseases and disorders:
- Cancer:
- Cytoskeletal proteins are often mutated in cancer, leading to increased cell motility, invasion, and metastasis. Changes in actin dynamics enable cancer cells to migrate and invade other tissues.
- Neurodegenerative Diseases:
- Disorders like Alzheimer’s disease involve tau protein abnormalities, leading to the destabilization of microtubules in neurons.
- Parkinson’s and Huntington’s diseases are also associated with cytoskeletal dysfunction, affecting cellular transport and stability in neurons.
- Muscular Dystrophy:
- Mutations in proteins linking the cytoskeleton to the cell membrane, such as dystrophin, lead to weakened muscle cells and muscular dystrophy.
- Laminopathies:
- Mutations in nuclear lamins (intermediate filaments) cause diseases like Hutchinson-Gilford Progeria Syndrome, leading to premature aging due to defects in nuclear structure and genome organization.
- Immune Disorders:
- Cytoskeletal defects can impair immune cell functions, such as migration, phagocytosis, and response to pathogens, leading to compromised immune responses.
Nucleus
The nucleus is a membrane-bound organelle found in eukaryotic cells that serves as the control center, housing the cell’s genetic material (DNA) and coordinating activities such as growth, metabolism, and reproduction. It plays a central role in gene expression, DNA replication, and cell division, making it essential for cellular function and organismal development. The nucleus is typically the most prominent organelle in the cell and contains various substructures that contribute to its functions.
Structure of the Nucleus
- Nuclear Envelope:
- The nuclear envelope is a double-membrane structure that surrounds the nucleus, separating it from the cytoplasm.
- The outer membrane is continuous with the endoplasmic reticulum (ER), allowing for connections between the nucleus and ER.
- The inner membrane provides structural support and is attached to the nuclear lamina.
- Nuclear Pores: Large protein complexes embedded in the nuclear envelope that control the passage of molecules, such as RNA and proteins, between the nucleus and cytoplasm. Nuclear pores allow selective transport, ensuring that only specific molecules can enter or leave the nucleus.
- Nuclear Lamina:
- The nuclear lamina is a network of intermediate filament proteins (lamins) just beneath the inner membrane. It provides structural support to the nuclear envelope and maintains the shape of the nucleus.
- The lamina also plays a role in organizing chromatin and regulating DNA replication and gene expression.
- Nucleoplasm:
- The nucleoplasm is the semi-fluid substance inside the nucleus, similar to the cytoplasm of the cell, where various molecules like nucleotides, enzymes, and ions are dissolved.
- It contains the chromatin and nucleolus, as well as enzymes involved in nuclear processes such as transcription and replication.
- Chromatin:
- Chromatin is the complex of DNA and proteins (mainly histones) that packages DNA into a more compact, organized structure.
- Chromatin exists in two forms: euchromatin (less condensed, transcriptionally active) and heterochromatin (more condensed, transcriptionally inactive).
- During cell division, chromatin condenses further to form visible chromosomes.
- Nucleolus:
- The nucleolus is a dense, spherical region within the nucleus that is not membrane-bound.
- It is the site of ribosomal RNA (rRNA) synthesis and ribosome assembly.
- Ribosomal subunits are assembled in the nucleolus, then transported to the cytoplasm through nuclear pores, where they combine to form functional ribosomes.
Functions of the Nucleus
- Storage and Protection of Genetic Material:
- The nucleus stores the cell’s DNA, which contains the instructions for building proteins and regulating cellular functions.
- By isolating the DNA within a membrane, the nucleus protects it from potentially damaging processes in the cytoplasm.
- Gene Expression and Regulation:
- The nucleus controls gene expression by regulating transcription (the process by which RNA is synthesized from DNA).
- Proteins called transcription factors and other regulatory molecules influence which genes are turned on or off, allowing the cell to respond to internal and external signals.
- RNA processing, including splicing, modification, and export, occurs in the nucleus, allowing for the production of mature messenger RNA (mRNA) that carries genetic information to ribosomes for protein synthesis.
- DNA Replication and Cell Division:
- Before a cell divides, its DNA is duplicated through DNA replication, ensuring that each daughter cell receives an identical set of genetic information.
- The nucleus organizes and coordinates DNA replication, as well as the distribution of chromosomes during mitosis and meiosis.
- Ribosome Biogenesis:
- The nucleolus within the nucleus is responsible for synthesizing rRNA and assembling ribosomal subunits.
- These subunits are then exported to the cytoplasm, where they participate in translating mRNA into proteins.
- Nuclear Transport:
- The nuclear envelope regulates the import and export of molecules via nuclear pores.
- Proteins, RNA molecules, and ribosomal subunits are actively transported through the nuclear pores with the assistance of transport proteins and signals.
Nuclear Processes
- Transcription:
- Transcription is the process by which an mRNA molecule is synthesized from a DNA template, using RNA polymerase enzymes.
- During transcription, only specific genes are transcribed, depending on the cell type, environmental conditions, and other regulatory signals.
- RNA Processing:
- Once mRNA is synthesized, it undergoes processing before leaving the nucleus, including splicing (removing introns), 5′ capping, and 3′ polyadenylation.
- Processed mRNA is then exported through nuclear pores into the cytoplasm for translation.
- DNA Replication:
- During the S phase of the cell cycle, DNA is replicated to produce identical copies of each chromosome, ensuring genetic continuity.
- Replication machinery within the nucleus unwinds and copies the DNA, producing sister chromatids that will later be separated during mitosis or meiosis.
- Cell Cycle Regulation:
- The nucleus contains proteins and checkpoints that regulate the cell cycle, ensuring that DNA is correctly replicated and any DNA damage is repaired before cell division.
- Failure in cell cycle regulation can lead to uncontrolled cell division, often associated with cancer.
- Chromatin Organization and Epigenetics:
- The nucleus organizes chromatin into specific regions that influence gene accessibility and expression.
- Epigenetic modifications, such as DNA methylation and histone modification, can alter gene expression without changing the DNA sequence, playing a role in cellular differentiation, development, and response to environmental factors.
Nuclear Disorders and Diseases
Malfunctions in nuclear structure or function can lead to various diseases, including:
- Cancer:
- Cancer often involves mutations in genes that regulate the cell cycle, leading to uncontrolled cell division.
- Alterations in nuclear architecture, DNA repair mechanisms, and chromatin organization are common in cancer cells.
- Genetic Disorders:
- Mutations in nuclear lamina proteins (lamins) can lead to disorders such as progeria, which causes premature aging.
- Defects in DNA repair genes increase susceptibility to genetic mutations and diseases, including certain cancers.
- Neurodegenerative Diseases:
- Mutations affecting nuclear proteins or chromatin structure can contribute to neurodegenerative disorders like Huntington’s and Alzheimer’s disease, impacting neuron function and survival.
- Muscle Diseases (Laminopathies):
- Diseases such as Emery-Dreifuss muscular dystrophy and limb-girdle muscular dystrophy result from mutations in nuclear lamina proteins, affecting muscle cell function and structure.
References
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2002). Molecular Biology of the Cell (4th ed.). New York: Garland Science.
- Lodish, H., Berk, A., Kaiser, C. A., Krieger, M., Bretscher, A., Ploegh, H., & Matsudaira, P. (2008). Molecular Cell Biology (6th ed.). New York: W.H. Freeman.
- Campbell, N. A., & Reece, J. B. (2005). Biology (7th ed.). San Francisco: Benjamin Cummings.
- Cooper, G. M., & Hausman, R. E. (2013). The Cell: A Molecular Approach (6th ed.). Sinauer Associates.
- Alberts, B., Bray, D., Hopkin, K., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2013). Essential Cell Biology (4th ed.). New York: Garland Science.
- Karp, G. (2010). Cell and Molecular Biology: Concepts and Experiments (6th ed.). Hoboken, NJ: Wiley.
- Pollard, T. D., Earnshaw, W. C., Lippincott-Schwartz, J., & Johnson, G. T. (2017). Cell Biology (3rd ed.). Elsevier.
- Hardin, J., Bertoni, G., & Kleinsmith, L. J. (2012). Becker’s World of the Cell (8th ed.). Boston: Pearson.